Chemprop: Aggregation and Extrapolation
Context
I have just finished a first version of my Master’s Thesis and waiting for my supervisor’s opinion. That’s why I again have tones of time for reading my favorite topics and seek some junior job position (😭). Recently, I came across Dr. Pat Walters’s blogs about the extrapolation capacity of Chemprop model.
Part 1: https://patwalters.github.io/Why-Dont-Machine-Learning-Models-Extrapolate/
Part 2 (guest post from Dr. Alan Cheng and Jeffery Zhou): https://patwalters.github.io/GNNs-Can-Extrapolate/
In Part 1, Dr. Walters suggested an interesting point: Chemprop models struggle with the extrapolation of Molecular Weights (MW). Specifically, models trained on compounds with MW below 400 g/mol have difficulty making predictions for those with MW higher than 500 g/mol. I refer to this as extrapolation on the target space, since it does not involve the input space, such as molecule clusters or chemical spaces, in this context.
In the follow-up blog, Dr. Cheng and Zhou raised an even more interesting point: using Mean Aggregation is the reason why model struggles with extrapolating MW. They proposed that using Norm Aggregation could greatly enhance extrapolation for MW. Based on their scripts, I recreated the analysis.

I then gave Sum Aggregation a try, which also performed well for MW extrapolation, as I expected. Because I noticed that MW is an additive property, it makes both Sum Aggregation and Norm Aggregation naturally advantageous.
This led me to question whether non-additive molecular properties can also benefit from Sum Aggregation or Norm Aggregation for extrapolation. This motivated me to write this blog. The focus of this blog is on the effect of aggregation layer on the Chemprop models’ extrapolation capacity. I used the default hyperparameters for the models in my analysis. Further investigation into hyperparameter tuning and extrapolation concerning the input space, such as scaffold or cluster-based splitting, would be out of scope but worth to try (maybe another blog).
The scripts for this blog is on my GitHub. Those scripts are mainly inherited from Jeffery notebook with a little modification to conduct my analysis.
Chemprop Aggregation Layer
Chemprop is a nice package for molecular property prediction. The fundamental architecture of Chemprop include 4 modules: (1) a local feature encoder to turn atoms and bonds into matrix representations, (2) a Directed Message Passing Neural Network (D-MPNN) to capture the relationship between atoms and bonds, (3) an aggregation function to combine atomic embedding into the molecular embedding, and (4) a standard FFN to generate final prediction from molecular embedding.
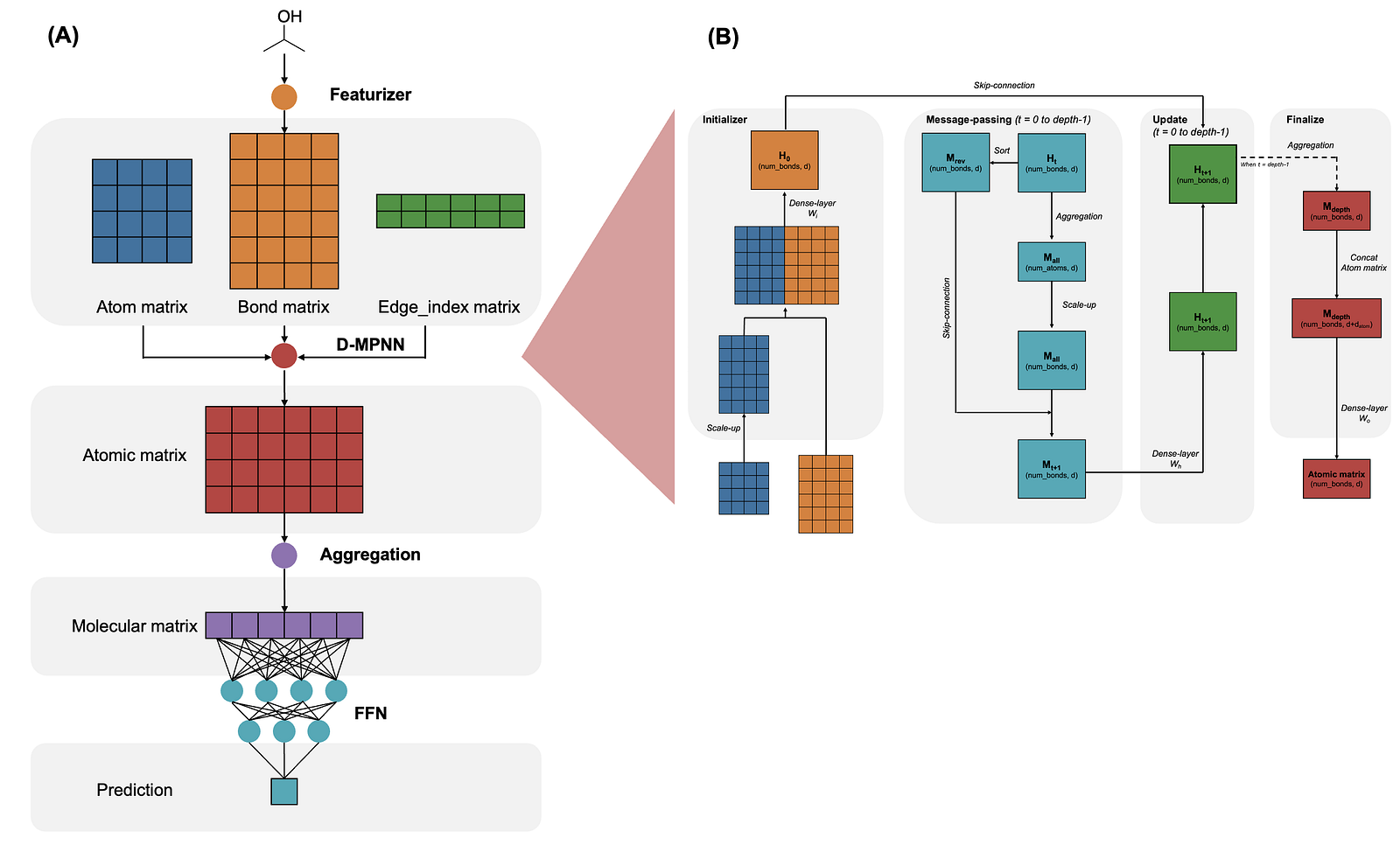
Recently, Chemprop supports 4 aggregation functions:
- Sum Aggregation: simply sums the atom matrix along the second dimension (the dimension of number of atoms).
- Norm Aggregation: performs sum aggregation and then divides the result by a constant norm factor, i.e. 100.
- Mean Aggregation: performs sum aggregation and then divides the result the number of atoms in the given molecule.
- Attentive Aggregation: assign attention scores for each atom using a learnable linear layer, then performs a weighted sum of atom features based on these attention scores, giving more importance to certain atoms in the aggregation.
Experiments
I applied the same approach to Dr. Walters, which consists of 3 datasets for each experiment: training-validating set, interpolating testing set, and extrapolating testing set. In my investigation, I examined four aggregation layers and seven target values: molecular weight (MW), logP, norm_MW (calculated as MW/number of atoms), TPSA, BalabanJ, BertzCT, and LabuteASA. I intentionally computed norm_MW as a way to reduce size-dependent property of MW.
Diagnostic plots were created for the testing sets, with the interpolating set represented in blue and the extrapolating set in orange.

To clarify, among the seven target values provided, MW, logP, TPSA, BertzCT, and LabuteASA are dependent on molecular size, while norm_MW and BalabanJ are size-independent. It is important to emphasize that my classification is based on how RDKit calculates these values, rather than their nature, as I derived these target values using RDKit.
There is an interesting trend: while Norm and Sum Aggregation perform well for extrapolating size-dependent properties, they fail for size-independent factors. Interestingly, Attentive and Mean Aggregation are effective for extrapolating size-independent properties but not for size-dependent factors. Norm_MW is a challenging task in general and might require hyperparameter tuning to boost the performance. However, looking at those that are highly correlated with y_true (points close to the diagonal line), Attentive and Mean Aggregation still perform better than the other two in terms of extrapolation.
Final thoughts
This blog shows an intuitive perspective that the selection of Aggregation layers is task-specific and worth for a very deep investigation when start training a Chemprop model. To strongly conclude, I believe that my toy models (I borrow this term from Dr. Walters) are not enough, as it might need a stronger statistical framework to draw the final conclusion.



Comments
Post a Comment