Data duplication definition is flexible. How can we handle it?
Duplication or not duplication?
I’m currently enjoying an extended holiday in Sweden, from Easter to Valborg, which gives me plenty of time to relax and read my favorite topic. Recently, I came across an insightful review article by Srijit Seal et al. A particular section of it reminded me of the many mistakes I encountered when I first began my cheminformatics journey, which is data leakage. While the article focuses on machine learning for toxicology prediction, the insights and warnings it offers are applicable to improving QSAR modeling in general. This inspired me to write a blog discussing both the obvious and hidden data leakage, as well as my approach to addressing the issue.
Data leakage — the devil
Data leakage is a real devil in machine learning training protocols. There are many forms of data leakage, but one of the most common problems arises when training data overlaps with validation data. In other words, this occurs when there are duplicates present in both the training and validation sets, which can lead to artificially optimistic performance metrics.
This problem is particularly prevalent when training machine learning models for small-molecule drugs, as the same compound can be represented in multiple ways. I will call this situation obvious duplication. For example, the two different SMILES codes below represent the same compound, acetyl salicylic acid.
SMILES_1: CC(=O)OC1=CC=CC=C1C(=O)O
SMILES_2: O=C(O)C1=CC=CC=C1OC(C)=O 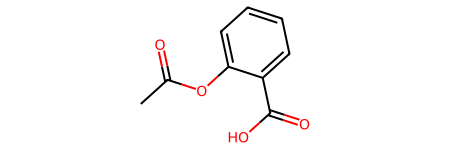
The dangerous part about data leakage is that it can occur naturally and go unnoticed. Researchers often use data compiled from various sources or public databases, where the same compound might be represented in different formats of SMILES strings. Without careful checks, these variants can end up split across the training and validation sets, unintentionally introducing data leakage. Therefore, it is crucial to control for duplicates and ensure proper deduplication in the dataset to prevent data leakage.
Duplications are domain-specific - Hidden duplication
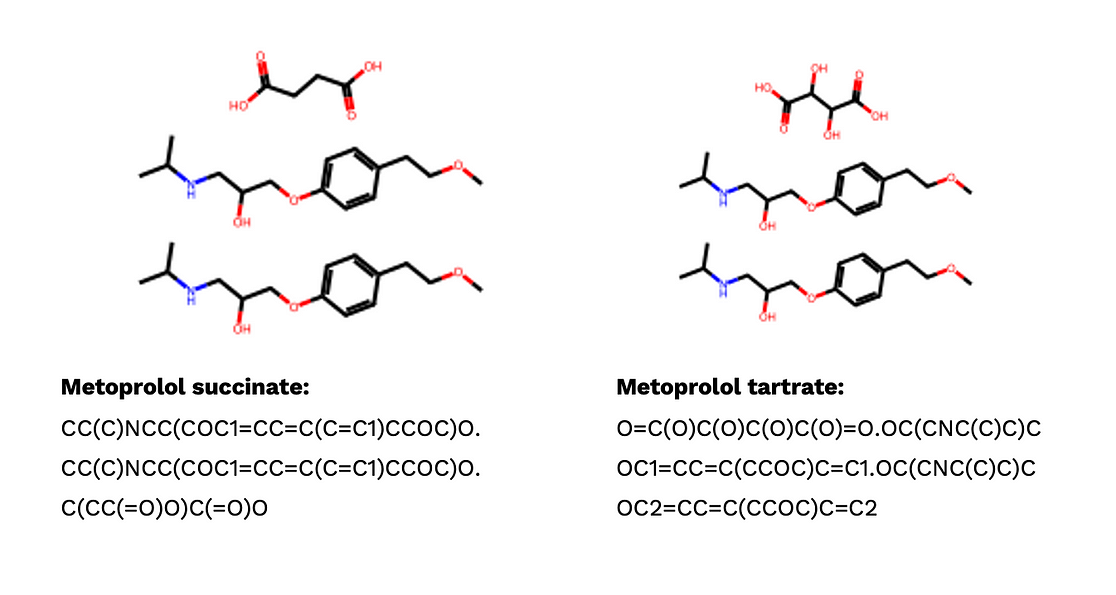
Beyond different SMILES strings for one compound, the real challenge of de-duplication in cheminformatics is that two entities may be considered duplicates or distinct depending on the specific domain context. Let’s have a look at these two salts:
When focusing solely on the active pharmaceutical ingredient (API), which in this case is metoprolol, these two salts could be viewed as duplicates. Metoprolol is known to block beta-1 adrenergic receptors, leading to a reduction in heart rate and potentially aiding in the treatment of hypertension. However, from a pharmacokinetic perspective, they should be considered distinct. This distinction arises from differences in formulation, crystal structure, solubility, dissociation rates, release rates, and other factors.
Therefore, if we develop machine learning models to predict the binding affinity of metoprolol to a specific target, we should eliminate one of the salts to prevent data leakage. Conversely, if we want to investigate how formulation impacts the release rate, both salts should be retained. In general, if the aspect we are considering, such as salt formation, is relevant to the model’s outcome, we should treat it as distinct when dealing with duplications. Conversely, if it is not relevant, we should consider it a duplication.
The salt formulation is one example to illustrate the domain-specific property of duplication. In reality, we have many more aspects to consider. They could be protonated state, stereochemistry, tautomers, and so on. From my perspective, we need a flexible standardization of SMILES strings, which can flexibly turn on neutralized protonated state, ignore stereochemistry, or standardize tautomers. To decide which function to turn on, it again depends on your specific problems.
De-duplication for obvious duplication
One of the simplest ways to de-duplicate compounds is to generate InChI for them, then drop duplication based on InChI. It is because, unlike SMILES, InChI is unique for each compound. This approach can work well with the obvious duplication; however, unable to handle hidden duplication.
from rdkit import Chem
# Obvious Duplications
smiles_1 = 'CC(=O)OC1=CC=CC=C1C(=O)O'
smiles_2 = 'O=C(O)C1=CC=CC=C1OC(C)=O'
print('Preprocessed SMILES')
print(f'SMILES 1: {smiles_1}')
print(f'SMILES 1: {smiles_2}\n')
mol_1 = Chem.MolFromSmiles(smiles_1)
inchi_1 = Chem.MolToInchi(mol_1)
mol_2 = Chem.MolFromSmiles(smiles_2)
inchi_2 = Chem.MolToInchi(mol_2)
print('InChI')
print(f'InChI 1: {inchi_1}')
print(f'InChI 2: {inchi_2}')
print(f'Equivalent tested: {inchi_1==inchi_2}') Preprocessed SMILES
SMILES 1: CC(=O)OC1=CC=CC=C1C(=O)O
SMILES 1: O=C(O)C1=CC=CC=C1OC(C)=O
InChI:
InChI 1: InChI=1S/C9H8O4/c1-6(10)13-8-5-3-2-4-7(8)9(11)12/h2-5H,1H3,(H,11,12)
InChI 2: InChI=1S/C9H8O4/c1-6(10)13-8-5-3-2-4-7(8)9(11)12/h2-5H,1H3,(H,11,12)
Equivalent tested: True
One similar approach is to generate canonical SMILES strings from the initial SMILES. Canonical SMILES strings use a “canonicalization” algorithm to reorder atoms and bonds in the molecular graphs in a consistent way.
from rdkit import Chem
# Obvious Duplications
smiles_1 = 'CC(=O)OC1=CC=CC=C1C(=O)O'
smiles_2 = 'O=C(O)C1=CC=CC=C1OC(C)=O'
print('Preprocessed SMILES')
print(f'SMILES 1: {smiles_1}')
print(f'SMILES 1: {smiles_2}\n')
mol_1 = Chem.MolFromSmiles(smiles_1)
canonical_smiles_1 = Chem.MolToSmiles(mol_1, canonical=True)
mol_2 = Chem.MolFromSmiles(smiles_2)
canonical_smiles_2 = Chem.MolToSmiles(mol_2, canonical=True)
print('Canonical SMILES')
print(f'Canonical SMILES 1: {canonical_smiles_1}')
print(f'Canonical SMILES 2: {canonical_smiles_2}')
print(f'Equivalent tested: {canonical_smiles_1==canonical_smiles_2}')Preprocessed SMILES
SMILES 1: CC(=O)OC1=CC=CC=C1C(=O)O
SMILES 1: O=C(O)C1=CC=CC=C1OC(C)=O
Canonical SMILES
Canonical SMILES 1: CC(=O)Oc1ccccc1C(=O)O
Canonical SMILES 2: CC(=O)Oc1ccccc1C(=O)O
Equivalent tested: True
De-duplication for hidden duplication
As I mentioned above, the hidden duplication is domain-specific, hence, we need flexible tools to handle SMILES strings. MolVs is well-known for its SMILES standardizer pipeline. MolVs standardize function offers 5 steps:
Step 1: Remove explicit hydrogen from molecular graphs.
Step 2: Disconnect metals.
Step 3: Normalize functional groups.
Step 4: Reionize functional groups according to pKa order.
Step 5: Remove symmetric stereocenters.
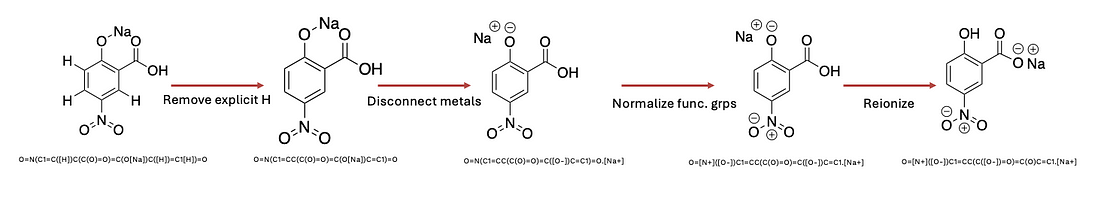
It is worth mentioning that molvs.Standardizer().standardize has nothing to do with removing salt, neutralizing functional groups, removing stereochemistry, and standardizing tautomers. If we have two tautomers, applying molvs.Standardizer().standardize still returns two different SMILES strings.
from molvs import Standardizer
smiles_1 = 'OC1=CC=NC=C1'
smiles_2 = 'O=C1C=CNC=C1'
for smiles in [smiles_1,smiles_2]:
mol = Chem.MolFromSmiles(smiles)
std_mol = Standardizer().standardize(mol)
std_smiles = Chem.MolToSmiles(std_mol)
print(f"Original: {smiles}")
print(f"Standardized: {std_smiles}")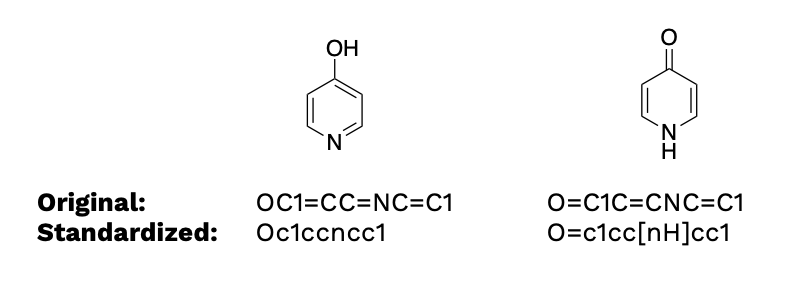
Instead of simply applying the molvs.Standardizer().standardize, I recommend having a look at their *_parents methods, which provide more robust functions to desalt, remove isotope, remove stereochemistry, remove charge, and standardize tautomers. They are offered by five wonderful methods in the Standardizer class:
from molvs import Standardizer
Standardizer().tautomer_parent(mol, skip_standardization = False)
Standardizer().fragment_parent(mol, skip_standardization = False)
Standardizer().stereo_parent(mol, skip_standardization = False)
Standardizer().isotope_parent(mol, skip_standardization = False)
Standardizer().charge_parent(mol, skip_standardization = False)By using these five methods plus the Standardizer() standardize (mol), we can flexibly establish our pipeline to de-duplicate with hidden duplication. For example, let's say I established a model that has nothing to do with salt, or two organic anions with different counter ions could be considered as the same. Thus, I want to focus on the largest fragment in the molecule, which is the active ingredient only. I would establish something like:
from molvs import Standardizer
class SMILES_standardize:
def __init__(self, smiles):
self.smiles = smiles
self.mol = Chem.MolFromSmiles(smiles)
Chem.SanitizeMol(self.mol)
def additional_standardizer(self):
# This is specific to your problem
self.mol = Standardizer().fragment_parent(self.mol, skip_standardize=False)
#self.mol = Standardizer().isotope_parent(self.mol, skip_standardize=False)
#self.mol = Standardizer().charge_parent(self.mol, skip_standardize=False)
#self.mol = Standardizer().stereo_parent(self.mol, skip_standardize=False)
#self.mol = Standardizer().tautomer_parent(self.mol, skip_standardize=False)Final thoughts
Data leakage and the definition of duplication can vary significantly across different domains, particularly in cheminformatics. This variability necessitates a flexible approach to determining what factors contribute to the definition of duplication in order to prevent data leakage. As a result, establishing a universal pipeline for handling SMILES strings is quite challenging.
This blog highlights the importance of addressing duplication in cheminformatics datasets and acknowledges its domain-specific nature. Recently, I implemented a solution using the MolVS library. However, there may be simpler, faster, and more optimal methods for standardizing SMILES. I encourage you to share any suggestions or insights you might have in the comments below.


Comments
Post a Comment